Apa perbedaan antara alkaptonuria dan phenylketonuria
Itu perbedaan utama Antara Alkaptonuria dan Phenylketonuria adalah bahwa alkaptonuria adalah kelainan genetik yang diwariskan yang dihasilkan dari ketidakmampuan untuk memetabolisme dua asam amino, tirosin dan fenilalanin, sedangkan fenilketonuria adalah gangguan genetik yang diwariskan yang dihasilkan dari ketidakmampuan untuk memetabolisme asam amino fenilalanin.
Kesalahan metabolisme bawaan adalah gangguan genetik yang diwariskan jarang. Dalam kondisi ini, tubuh tidak dapat dengan benar mengubah makanan menjadi energi. Gangguan kesalahan metabolisme bawaan biasanya disebabkan karena cacat pada enzim spesifik yang membantu memecah bagian makanan. Ada banyak jenis kesalahan metabolisme bawaan. Beberapa dari mereka adalah intoleransi fruktosa, galaktosemia, penyakit urin gula maple, alkaptonuria, dan fenilketonuria.
ISI
1. Ikhtisar dan Perbedaan Utama
2. Apa itu Alkaptonuria
3. Apa itu fenilketonuria
4. Kesamaan -Alkaptonuria dan Phenylketonuria
5. Alkaptonuria vs Phenylketonuria dalam bentuk tabel
6. Ringkasan -Alkaptonuria vs Phenylketonuria
Apa itu Alkaptonuria?
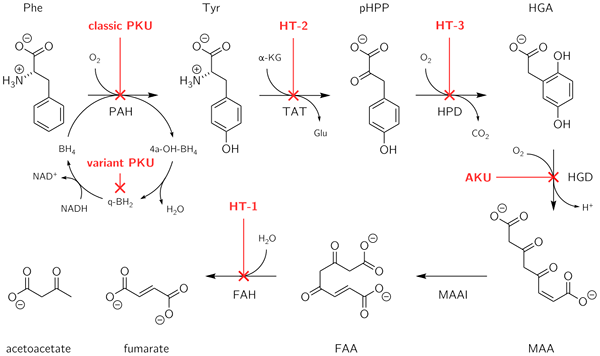
Alkaptonuria adalah kelainan genetik yang diwariskan yang dihasilkan dari ketidakmampuan untuk memetabolisme dua asam amino: tirosin dan fenilalanin. Ini adalah jenis kesalahan metabolisme bawaan. Gangguan ini disebabkan oleh mutasi pada gen yang disebut HGD, yang bertanggung jawab untuk membuat enzim yang dikenal sebagai HGD (Homogentisate 1, 2-Dioxygenase). Enzim HGD terlibat dalam metabolisme asam amino aromatik tirosin dan fenilalanin. Orang yang memiliki mutasi HGD tidak dapat memetabolisme asam homogentisat yang dihasilkan dari tirosin menjadi 4-maleylacetoacetate. Cacat ini menghasilkan akumulasi asam homogentisik dalam darah dan jaringan. Selain itu, dalam kondisi ini, asam homogentisik dan bentuk teroksidasi (alkapton) diekskresikan dalam urin, yang memberikan warna gelap yang luar biasa untuk urin.
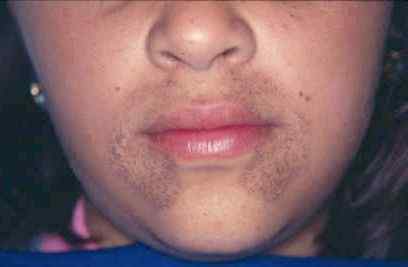
Gambar 01: Alkaptonuria
Tanda dan gejalanya termasuk jaringan bernoda gelap di dalam tubuh, masalah sendi dan tulang (osteoartritis), penebalan dan perubahan warna hitam tulang rawan telinga (ochronosis), lilin telinga hitam atau kemerahan, bintik-bintik coklat atau abu-abu di atas putih dari putih Mata, keringat yang berubah warna, area kulit berwarna biru atau hitam, kuku warna kebiruan, kesulitan bernapas, masalah jantung (katup jantung yang mengeras dan pembuluh darah yang kaku), batu ginjal, batu kandung kemih, dan batu prostat. Diagnosis kondisi ini dapat dilakukan melalui pemeriksaan fisik, riwayat pasien terperinci, uji urin, kromatografi gas untuk menguji jejak asam homogentisik, dan uji DNA untuk memeriksa mutasi gen HGD. Selain itu, pilihan pengobatan termasuk memberikan nisiton untuk mengurangi asam homogentisik dalam tubuh, membatasi protein dalam diet, olahraga untuk rasa sakit dan kekakuan, dan menghilangkan rasa sakit melalui pembunuh rasa sakit.
Apa itu fenilketonuria?
Phenylketonuria adalah kelainan genetik yang diwariskan yang dihasilkan dari ketidakmampuan untuk memetabolisme hanya satu asam amino: fenilalanin. Kondisi ini biasanya karena mutasi pada gen yang dikenal sebagai PAH, yang mengkodekan enzim yang dikenal sebagai fenilalanin hidroksilase. Enzim ini diperlukan untuk memetabolisme fenilalanin asam amino menjadi asam amino tirosin. Ketika aktivitas fenilalanin hidroksilase berkurang karena mutasi, fenilalanin menumpuk dan dikonversi menjadi fenilpiruvat (fenilketon), yang dapat dideteksi dalam urin.
Gambar 02: Phenylketonuria
Tanda dan gejala gangguan ini mungkin termasuk bau apak pada keringat, kulit, atau urin, masalah neurologis termasuk kejang, kulit adil dan mata biru, kepala kecil yang tidak normal, hiperaktif, kecacatan intelektual, perkembangan yang tertunda, perilaku, emosi, masalah sosial, dan gangguan kejiwaan. Diagnosis dilakukan melalui pengujian darah yang baru lahir, evaluasi klinis, dan pengujian DNA untuk mutasi gen. Selain itu, pengobatan ini terutama melalui regulasi diet, yang meliputi makanan yang mengandung kadar fenilalanin rendah (membatasi protein) dan formula khusus untuk bayi dengan sejumlah kecil ASI ASI. Obat sapropterin dihydrochloride juga dapat berguna dalam beberapa kasus. Obat ini adalah kofaktor untuk enzim fenilalanin hidroksilase, yang meningkatkan aktivitasnya.
Apa kesamaan antara alkaptonuria dan fenilketonuria?
- Alkaptonuria dan phenylketonuria adalah dua kesalahan metabolisme bawaan.
- Keduanya diwariskan gangguan genetik.
- Gangguan ini mengikuti warisan resesif autosomal.
- Kedua gangguan tersebut menyebabkan akumulasi metabolit dalam jaringan tubuh.
- Mereka terutama diperlakukan dengan membatasi diet berbasis protein tinggi.
Apa perbedaan antara alkaptonuria dan phenylketonuria?
Alkaptonuria adalah kelainan genetik yang diwariskan yang dihasilkan dari ketidakmampuan untuk memetabolisme dua asam amino, tirosin dan fenilalanin, sedangkan fenilketonuria adalah gangguan genetik yang diwariskan yang dihasilkan dari ketidakmampuan untuk memetabolisme asam amino fenilalanine. Dengan demikian, ini adalah perbedaan utama antara alkaptonuria dan phenylketonuria. Selanjutnya, prevalensi global alkaptonuria adalah 1 pada 250000 hingga 1000000 kelahiran hidup, sedangkan prevalensi fenilketonuria global adalah 1 dalam 23930 kelahiran hidup.
Infografis di bawah ini menyajikan perbedaan antara alkaptonuria dan fenilketonuria dalam bentuk tabel untuk perbandingan berdampingan.
Ringkasan -Alkaptonuria vs Phenylketonuria
Kesalahan metabolisme bawaan adalah gangguan genetik yang diwariskan jarang. Alkaptonuria dan phenylketonuria adalah dua kesalahan metabolisme bawaan. Alkaptonuria dihasilkan dari ketidakmampuan untuk memetabolisme dua asam amino tirosin dan fenilalanin sementara fenilketonuria dihasilkan dari ketidakmampuan untuk memetabolisme fenilalanin asam amino. Jadi, ini merangkum perbedaan antara Alkaptonuria dan Phenylketonuria.
Referensi:
1. “Alkaptonuria.Pilihan NHS, NHS.
2. “Phenylketonuria (PKU).”Mayo Clinic, Mayo Foundation for Medical Education and Research.
Gambar milik:
1. "Ochronosis" oleh Universidad CES - (CC oleh 3.0) Via Commons Wikimedia
2. “Kesalahan Metabolisme Fenilalanin dan Tyrosine” oleh Bradford Morris - karya sendiri (CC BY -SA 4.0) Via Commons Wikimedia